Data analysis strategy through metAlign eyes
The bottleneck in metabolomics is still data analysis. The analytical instrumentation has evolved. Experiments are now highly automated and can output huge amounts of raw data in a relatively short time. It is easy to get swamped in data. Therefore in a full-cost calculation of a metabolomics experiment (presuming adequate samples are already present), data analysis costs will in general by far outweigh the measuring costs and also be the rate-determining factor in reporting results.
Obviously, the simplest way of cutting back on data-analysis time is to make sure that the data are of excellent quality. Badly designed or performed experiments are an expensive waste of time. But still, even with ideal experiments you need a lot of time to extract the signals, compare data and identify compounds.
Currently there are 2 ways of doing this. One way is a targeted analysis of a predefined set of compounds. The other is untargeted analysis, which entails comparison of all signals over different measurements, finding the interesting differences and then trying to do identification. Free (and probably better than commercial) packages that help in this are, for example, metAlign (http://pubs.acs.org/doi/abs/10.1021/ac900036d), XCMS and mzMine. All three handle LCMS data with different mass precision. MetAlign, however, allows for the highest mass precision and also handles GCMS well. Most of the identification work is extra.
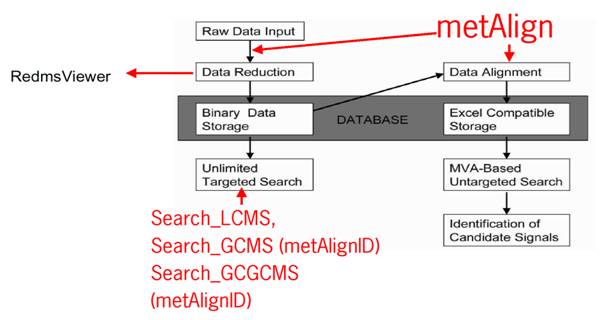
Figure 1: Scheme of metAlign and related software.
While XCMS for instance has gone towards a web application (http://pubs.acs.org/doi/abs/10.1021/ac300698c), metAlign will remain a local application. MetAlign 3.0 (http://link.springer.com/article/10.1007%2Fs11306-011-0369-1) is now ten times faster on a modern consumer PC (around 1000 Euros) than 2 years ago; it now can use all the processors of a PC. It therefore will scale with future consumer PC developments, unlike most other programs which still use one processor even if more are available. Fumio Matsuda at RIKEN is now running metAlign routinely on twenty-four cores on a 64-bit W7 PC (ten minutes for 2.1 GB of raw GC-MS data). The point is that metAlign is a good and very fast alternative to web applications and avoids the necessity of uploading huge amounts of data. It is easy to increase computing power and speed at a low cost and still be independent. An important thing remains, that import is flexible (different machines and formats) and that export can easily be used and built on.
Identification is still a very big challenge if no reference compound or entry in a spectral library is present. Interesting signals from untargeted analysis and multivariate analysis therefore have to be filtered or clustered as much as possible before identification can proceed. Based on metAlign 2 platforms are available, i.e., http://prime.psc.riken.jp/lcms/ and http://link.springer.com/article/10.1007/s11306-011-0368-2?null. Because of the high costs of absolute de novo identification (preparative isolation and additional full study with NMR is required), it is extremely important to have high quality signal leads. Checking leads is therefore required. Important developments here are GCxGC-TOF-MS for improved separation and cleaner mass spectra and high-resolution UPLC-MS or perhaps high-resolution GC-TOF-MS for more accurate mass and therefore better elemental composition analysis. For that matter, even LCxLC-MS is not that far away anymore as well as coupling GCxGC or LCxLC to high-resolution mass spectrometers. Post sub-ppm enhancement of mass accuracy for LC-Orbitrap-MS may help http://www.springerlink.com/content/1h1714h537p574t5/.
The trouble with GCxGC-TOF-MS and LCxLC-TOF-MS is that the data sets are very large and information rich. This adds tremendously to the data analysis work load. This seems to add up to a situation in which the increase in separation is out-balanced by the extra data handling. If high-resolution MS is added to this technology the data size will not only grow beyond what we can handle on a 32-bit operating system, but data-analysis time will again increase. The paradox is that it is technologically possible, but the data handling is not or extremely tedious.
So, again there is a great need to come up with solutions for speeding up data handling. And as with metAlign 3.0 the obvious thing is to scale up by using more processors on a PC. We realized this with metAlignID for high-throughput targeted analysis in GCxGC-TOF-MS (http://www.sciencedirect.com/science/article/pii/S0021967312014689). This tool was developed for routine residue and contaminant analysis in GCxGC-TOF-MS. Within one hour it is now possible to convert, pre-process and search 30 GCxGC-TOF-MS files for 560 compounds on a standard quad core consumer PC.
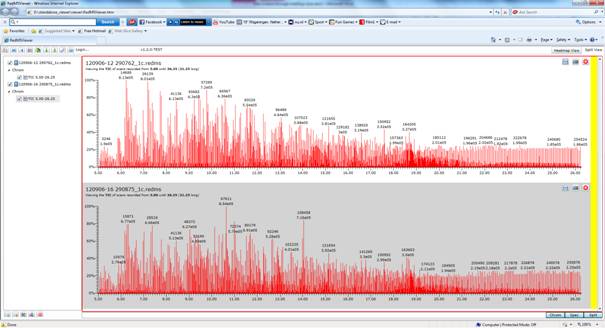
Figure 2: The redms viewer interface with two GCxGC-TOF-MS files loaded. This program can read in up to a 100 files due to their small size.
A nice feature of all the pre-processing is that the size of data files is decreased ca. 100-fold. If storage of data profiles in databases or transfer through internet is an issue, this will certainly help. We recently have developed a beta-version of a viewer, which is capable of displaying these small data files. The viewer and examples of pre-processed GCxGC-TOF files and a sub-ppm corrected file are in this link: https://dl.dropbox.com/u/75158272/viewer.zip
These small pre-processed files can not only be easily stored, but can also easily be searched in a fast way (see given metAlignID and sub-ppm reference) and will not give memory problems. Furthermore, the same small files (but not GCxGC-TOF) can be used for alignment and untargeted strategies as well as unlimited targeted searches.
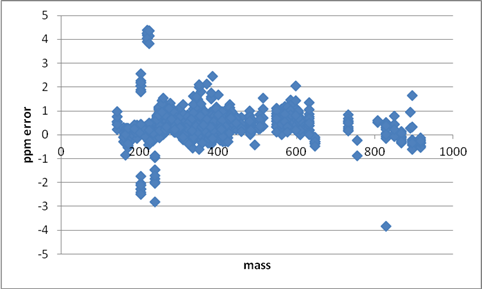
Figure 3: ppm error vs. mass of ten UHPLC Orbitrap in metAlign-processed data files (50000 resolution) after post-acquisition sub-ppm enhancement. 1535 experimental masses (including adducts) are obtained from a mix of 103 compounds at different concentrations. Outliers and errors in assignment are easily spotted.
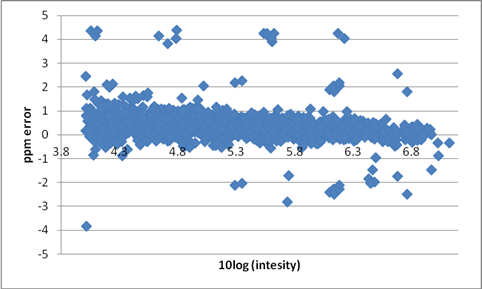
Figure 4: ppm error vs. 10log(intensity) of 10 UHPLC Orbitrap in metAlign-processed data files (50000 resolution) after post-acquisition sub-ppm enhancement. 1535 experimental masses (including adducts) are obtained from a mix of 103 compounds at different concentrations. Outliers and errors in assignment are easily spotted.
The current efforts based on metAlign output are going towards automation and combining sub-ppm enhancement of LC-Orbitrap-MS data (easier method and more routine), cleaning data for adducts, analysing isotope pattern information, elemental composition according to 7 golden rules ( http://www.biomedcentral.com/1471-2105/8/105/), fragment searches and analysis and coupling to databases. Perhaps MS fragment prediction will also come into play. One important aspect will also be quality of mass precision, which is resolution but also intensity dependent. The second is the accuracy/correction of the calculated number of carbons, which is resolution-, mass-, and intensity-dependent in Orbitrap data. For both these quality aspects we will be obtaining distributional information over hundreds to thousands of mass peaks using very fast searching programs.
Besides untargeted approaches focussing on extracting important signals, we believe that highly-automated instrumental screening (large scale targeted analysis) like in residue and contaminant analysis will become very important in making metabolomics more cost-effective.
Arjen Lommen